Computer Models Could Allow Researchers to Better Understand, Predict Adverse Drug Reactions
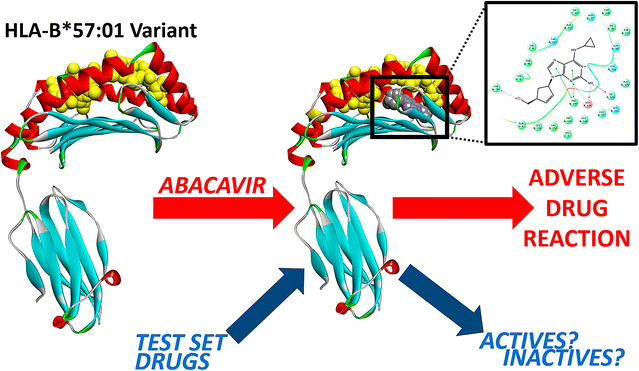
New computer models from North Carolina State University show how a variant of a common protein involved in human immune response binds to the antiviral drug abacavir, causing a severe life-threatening reaction known as the abacavir hypersensitivity syndrome (AHS). The work has implications for predicting severe adverse reactions caused by existing drugs and future drug candidates in subpopulations of patients.
Abacavir is a common anti-HIV drug. However, it is associated with severe allergic reactions in a fraction (5-8 percent) of patients who take it. Previous research determined that this response occurs in patients with a particular variant of a human leukocyte antigen (HLA) known as HLA-B*57:01.
Denis Fourches, assistant professor of chemistry at NC State, and his graduate student, George Van Den Driessche, wanted to know what happens at the molecular level when abacavir and other drugs interact with HLA-B*57:01.
HLA proteins reside on the surface of cells and help the immune system distinguish between its own proteins and those made by infectious agents. HLA proteins and their co-binding peptides serve as signaling proteins that communicate with T-cells, ensuring that all is well. If something foreign – a pathogen, or in this case, a drug like abacavir – binds to the HLA protein, displacing the co-binding peptide and deforming the overall shape of the molecular complex, this change is recognized by immune cells, triggering the immune response.
“There are 15,000 variants of HLA, and everyone carries some of these variants,” Fourches says. “HLA-B*57:01 is one of the first variants studied in the context of a drug-induced immune reaction. We know that it binds with abacavir, but little was understood about exactly what was happening structurally at the molecular level, especially in terms of the relationship with the co-binding peptide. This is a very complex system.”
Fourches and Van Den Driessche created a series of computer models using three-dimensional molecular docking that allowed them to look at the ways in which abacavir docks in the binding site of HLA-B*57:01 – where the co-binding peptide normally docks. The models incorporated 3D structures of abacavir, HLA-B*57:01, and several potential co-binding peptides. The researchers ran molecular modeling simulations to virtually dock abacavir in the HLA-B*57:01 active site in the absence and presence of a co-binding peptide. Finally, they virtually screened an additional set of 13 drugs, some of which are known to cause severe adverse responses that are also suspected of being linked to HLA variants.
“The models allowed us to identify key atomic interactions that cause abacavir and other drugs to bind to the HLA variant protein and ultimately trigger the immune response,” Fourches says. “When you can forecast and understand the elements of the drug that enable the binding to occur, you may be able to create new active compounds that do not have that problem.
“Our ultimate goal is to use molecular modeling to discover and understand how different drugs can interact with the immune system via direct HLA binding interaction, so that we can better predict the side effects of new drugs and design drugs with fewer side -effects. This is critical for the personalized medicine of tomorrow.”
The research appears in the Journal of Cheminformatics, and was funded by the NC State Chancellor’s Faculty Excellence Program. Van Den Driessche is first author of the paper, and Fourches is the corresponding author.
-peake-
Note to editors: An abstract of the paper follows.
“Adverse Drug Reactions Triggered by the Common HLA-B*57:01 Variant: A Molecular Docking Study”
DOI: 10.1186/s13321-017-0202-6
Authors: George Van Den Driessche and Denis Fourches, North Carolina State University
Published: Journal of Cheminformatics
Abstract:
Background: Human leukocyte antigen (HLA) surface proteins are directly involved in idiosyncratic adverse drug reactions. Herein, we present a structure-based analysis of the common HLA-B*57:01 variant known to be responsible for several HLA-linked adverse effects such as the abacavir hypersensitivity syndrome.
Method: First, we analyzed three X-ray crystal structures involving the HLA-B*57:01 protein variant, the anti-HIV drug abacavir, and different co-binding peptides present in the antigenbinding cleft. We superimposed the three complexes and showed that abacavir had no significant conformational variation whatever the co-binding peptide. Second, we self-docked abacavir in the HLA-B*57:01 antigen binding cleft with and without peptide using Glide. Third, we docked a small test set of 13 drugs with known ADRs and suspected HLA associations.
Results: In the presence of an endogenous co-binding peptide, we found a significant stabilization (~2 kcal/mol) of the docking scores and identified several modified abacavir-peptide interactions indicating that the peptide does play a role in stabilizing the HLA-abacavir complex. Next, our model was used to dock a test set of 13 drugs at HLA-B*57:01 and measured their predicted binding affinities. Drug-specific interactions were observed at the antigen-binding cleft and we were able to discriminate the compounds with known HLA-B*57:01 liability from inactives.
Conclusions: Overall, our study highlights the relevance of molecular docking for evaluating and analyzing complex HLA-drug interactions. This is particularly important for virtual drug screening over thousands of HLA variants as other experimental techniques (e.g., in vitro HTS) and computational approaches (e.g., molecular dynamics) are more time consuming and expensive to conduct. As the attention for drugs’ HLA liability is on the rise, we believe this work participates in encouraging the use of molecular modeling for reliably studying and predicting HLA-drug interactions.
This post was originally published in NC State News.